The NFL CTE Study a Walkthrough

Jim Harbaugh called out an Attack on Football in 2015. That rhetoric has subsided for the most part. There is however a new attack that literally came to a very Michigan head on Sept 6th in a Frank Bruni editorial in the New York Times (NYT) “Can We Talk About Tom Brady’s Brain?” This opinion piece focused on the general public’s avoidance of discussing Gisele Bundchen’s public statement that Brady had undisclosed concussions last season.

New York Daily Post linked from web
Last Friday a head of a different order was on the front page of the NYT as well.

Credit Pool photo by Steven Senne - NYT linked from web
Both articles focused on chronic traumatic encephalopathy (CTE) in cases that, in my opinion and for different reasons, more than likely had nothing to do with it. The frequency of these stories, especially in the NYT, is exceeded only by recent CTE posts to the board.
The media has misrepresented the state and content of CTE research. Much of the fan reaction has been either alarmist or off base. The actual science has taken a back seat to the public hysteria. The interests of non professional football players who stand at greatest risk if only by virtue of their shear numbers are in danger of losing the learning science has to offer by the lack of focus on what the actual studies are saying.
This post is a walk through of the most recent and comprehensive CTE study to date from the July 25th 2017 Journal of the American Medical Association (JAMA) entitled “Clinicopathological Evaluation of Chronic Traumatic Encephalopathy in Players of American Football”. Most of us don’t have time to dig these studies out and come to terms with them. This is an attempt to bring that data to those who wouldn’t otherwise look at it. If I have time there’s another study that came out this past week by Robert Stern that deserves reading as well.
The lead author of the first study is Ann McKee. The study represents the work of many scientists but in the discussion below I will refer to it by her last name.
Here is a timeline of CTE leading up to this most recent report.
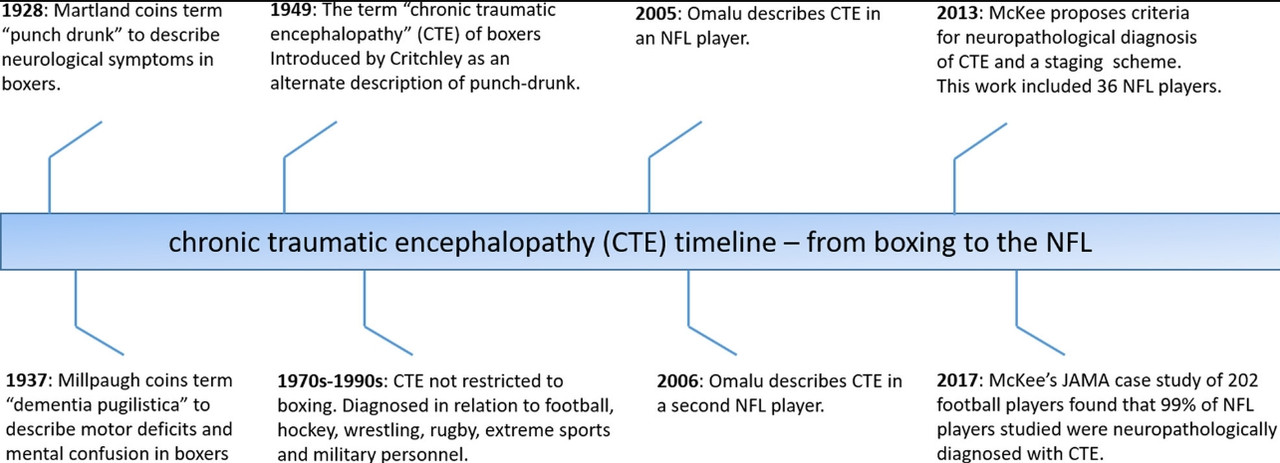
McKee has increased her dataset from 85 in 2013 to 202 players for this most recent publication. These 202 brains are what is termed a “convenience” sample. This is a new term (with respect to her 2013 write-up) and clarifies one of the primary public criticisms by non-scientific readers of McKee’s earlier work. The subjects of this study were all suspected of having a brain disorder prior to being accepted into the study. There is no control group here and the sampling is not random. The percent affected by CTE (80% in 2013 and 87% in the more recent study) doesn’t project to the general population of American football players. The increase in % affected from 2013 to this 2017 study doesn't necessarily reflect an increase in the incidence of CTE. Convenience is the correct term for this sampling. That understood – it’s not impossible to generalize – but first let’s get to what this study is saying before going there.
By far what is most convenient about McKee’s subjects is the fact that they are… dead. This is the biggest difference between concussion or mild traumatic brain injury (mTBI) and CTE research. The former is defined by symptoms and the later is defined by pathology post mortem. There is no conclusive test for CTE in the living. This means that there are no longitudinal studies of the subjects.
This is not to say there are no clinical symptoms to identify CTE. In fact there are no cases of CTE that are demonstrated without some behavioral or cognitive symptoms in the retrospective clinical evaluations. The problem is this… symptoms are often confounded by other neurodegenerative disorders. Actual confirmation of CTE is only possible by careful cross section and sample prep.
There are several other changes from 2013 in this new data. The informants were made to understand a more stringent definition of mTBI prior to submitting the intake evaluations. There are known issues in collecting accurate mTBI data. This extra step is to insure the data has a baseline expectation. I’m pretty sure this increases the mTBI numbers in this study. Football players have traditionally understated past mTBI in their life histories. That, I think, has probably changed but not significantly. Race was included as well. The criteria for inclusion was tightened due to cost and limited resource. Nineteen players were excluded due to both age (>35) and exposure (<2 years of college ball). All said only 36 brains from the 2013 study met the the new criteria.
What follows is the first table of data from McKee’s study.
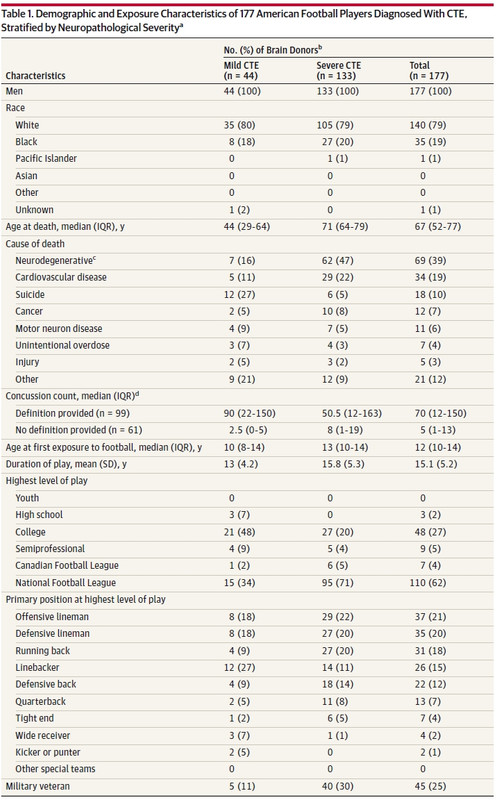
Though there is that new report by McKee’s fellow researcher Robert Stern last week (that I mentioned above) that focuses on youth football this isn’t that report. Very few high school and no youth players were included by design. The media’s front page treatment of McKee’s report does not provide younger players and their families with much data from which they can take informed action. Better disclaimers would clearly have helped the public.
Looking at this table one very real “inconvenient” bullet is pretty obvious as well (though strangely not discussed in the media or in McKee’s write up.) The distribution of the sample by race is 79% white 19% black and 2% other. The NFL population is nearly the exact opposite proportion (~70% black, 26% white and 4% other.) There’s no conspiracy here but the difference is not insignificant. It should be discussed.
There are four paths to getting brains into this study. Here’s a quick table showing these paths…
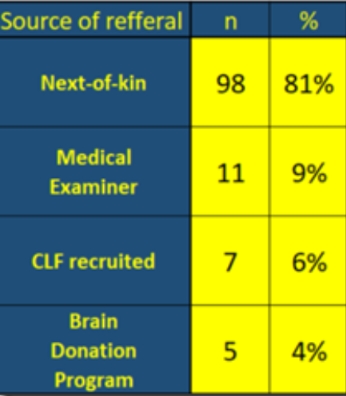
If there is a racial bias, and there is, it is in one or all of these paths. Recruitment is not broken down by race, however, in the study. This is a summary only since 2014 but it represents the current scope of this group’s recruitment strategy. CLF, the brain bank and the Brain Donation Program all refer to affiliated programs and are hyperlinked. Most of the subjects come from family members. It’s not hard to fathom why they would do this as these can be anonymous and it provides potential closure for people seeking answers about loved ones. It may seem odd that black families would not be as inclined to join the study in that light. Instead this is perhaps another example of differences in justifiable racial attitudes toward health science and medicine. Better outreach is clearly needed.
The reason this data was collected in the first place was to examine the difference in pathology . There is no rollup of that data other than in the table above. It looks matched across the two groups.
One last thing on race that brings out another potential source of error in this type of study. Football varies year to year, decade to decade by equipment, rules and coaching. This can present a cohort bias – 70s era players vs 90s era. The severe cases are on average 2 to 3 decades removed from the mild cases. Though the NFL is currently 80/20 black to white, the league was 30 /70 in the 70s (though I'm not seeing a summary of race/roster by year or decade.) Even with the 30/70 figure the race numbers are not reflective of players by era. More importantly the roughly equal sample by race between the younger/mild and older/severe subjects points to race bias in recruitment for this study.
The McKee study is 3 months old. It doesn’t talk to the Hernandez case directly (nor should it) but indirectly it does. Ann McKee went further last week and commented on the case speaking to the severity of Hernandez’ CTE. Speaking out on these lawsuits is not wise before they come to court. This is a powerful case in the public perception of CTE that science isn't ready to solve.
McKee does indirectly address the core issue of the Aaron Hernandez case in her discussion in the study however. I paraphrase here… 43% of the initial clinical intakes in this study showed mood and behavior symptoms on the initial intake presentation. Just as there are two different presentations (young with mood and behavior symptoms & old with cognitive issues) there are two paths Hernandez and CTE could have taken. CTE could be causative or contributory. The contributory model suggests that CTE could lower thresholds for psychiatric issues. McKee suggests that possibility in her discussion. The causative model would imply that Hernandez’ CTE would be more severe than other cases which is exactly what McKee commented last week. Looking at the slides McKee and others have published … they don’t look as severe as some older subjects. That judgment is McKee’s to make however and if you take the media’s report of what she said… McKee thinks Hernandez’ CTE was exceptionally severe given his age. If it is exceptional then the CTE study that came out in July doesn't have much to say about the causative model. The severest cases are generally older with cognitive deficits Hernandez didn’t apparently show. I guess there’s a third model … CTE doesn’t have much to do with murder. It will be interesting to see how that plays out.
Reading the actual papers gives insight sometimes but more often than not it creates more questions than answers. That is both a good thing and also science at it's best. Instead the media, the lawyers and the players are looking for the kind of answers that just aren't there in the science as yet.
OK… back to the study.
The mild vs. severe classification is determinant of a similar staging regime to that done in 2013. Stages I and II are considered mild, while Stage III and IV are severe. There has been quite a bit more work done by McKee and others on the codification of prep, diagnosis and classification of CTE since the previous study as well. McKee has a few slides that show these classifications.
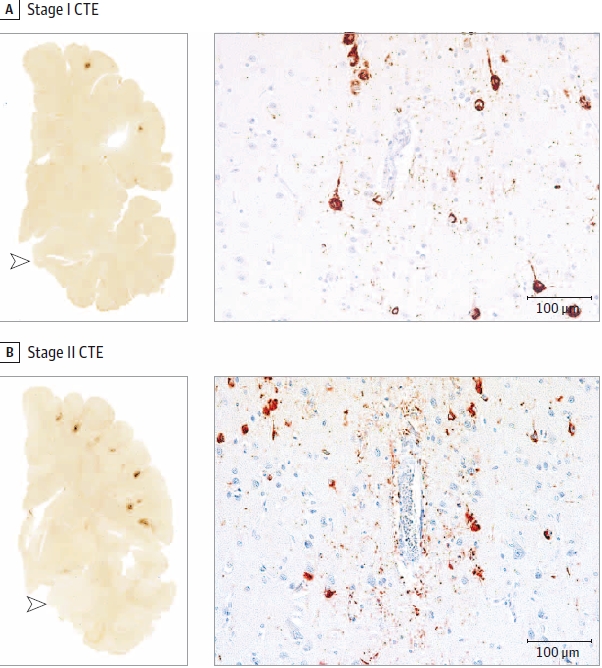
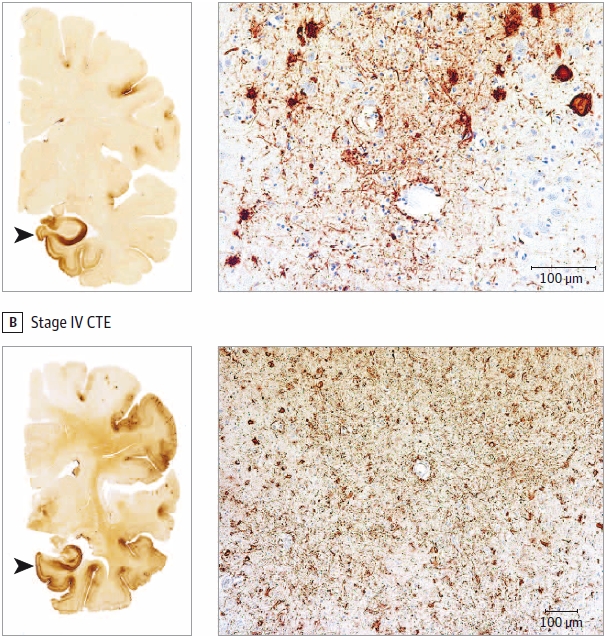
What are those dark regions? The methods used to get these slides highlight a p-tau protein that is unexpected for it’s location, density and frequency.
Here’s another digression since McKee only lightly touches on what p-tau is and for that matter what the model of CTE is… I am going to do that now. This is not in McKee or Stern’s study.
Tau proteins occur naturally in the axons of neurons. They actually occur in most cells. The “p” stands for phosphorylated… i.e. p-tau are tau proteins that have phosphate groups added. The tau protein is a stabilizing molecule within structures called microtubules on which your neurons transport neurotransmitters.
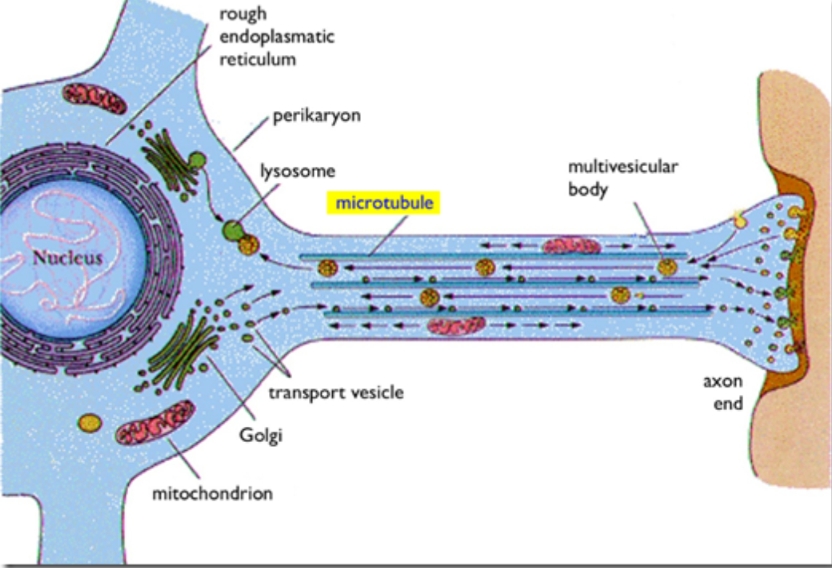
Why are microtubules and transport required? Because neurons are often one-sided affairs. They have a nucleus / cell body on one side and a long axon containing microtubules extending possibly very far away toward other neurons. The cell body produces critical molecules and neurotransmitters that the axon end or terminus can not manufacture locally. The microtubules are highways by which the cell transports these molecules. The scale of this is interesting and the classic pictures of motor neurons we are exposed to in high school don’t do it justice.
Make a fist. Imagine it is a neuronal cell body. Now sit down on the fifty yard line at Michigan stadium and grab a garden hose. The garden hose is the axon. To get to some of the more extreme axon terminals on the other end of the axon that garden hose would extend from your fist to the Diag. Inside of the axon would be microtubules on which are the stabilizing tau proteins. That is pretty a-maize-ing… to me at least. That is not true for all neurons but the scale on common figures like the one above, which is not a brain cell even, is a pedagogic conceit to show all structures and is grossly misleading.
The extreme dimensions of neurons are the biomechanical basis for the model of concussions being axonal damage due to shearing, rotational, pressure or stretching injury to axons. You can easily imagine these axons being susceptible to small movements especially across different geometries and tissue types of varying elasticity. The good news is they aren’t that fragile. However auto accidents, exposure to explosive devices and blunt trauma are challenges to the biomechanical stability of these garden hoses.
OK… digression is over… almost.
So what are those red dots in the images McKee is using to show examples of CTE staging? It turns out these tau proteins can disassociate from the microtubules, by disease or trauma, becoming p-tau through a process that is not well understood. These p-tau proteins can then form tangles called neurofibrillary tangles (NFTs) which is what is showing when McKee applies the correct stain and slide prep.

The figure above is taken from an Alzheimer Disease (AD) slide. These NFTs are fundamental to a model for AD. In fact p-tau and NFTs are often found in many forms of neurodegenerative disease. The scientists studying CTE are often AD researchers as well. McKee and Stern both can wear that hat. Some scientists have postulated that NFTs have a protective function in some scenarios. (Phosphorylation itself is a fundamental process in the control of mitosis in all cells that have a nucleus. It's not inherently a bad thing.) In CTE the NFTs cause cell death or neural degradation.
The model for NFT generation in CTE is not well understood. This is why concussive and non-concussive head trauma are usually indicated as precursors. We don’t know exactly if it’s trauma, inflammation from trauma or… you get the idea… we don’t conclusively know how CTE occurs. The causal relationship of CTE to TBI (concussive/mTBI or subconcussive) is based on correlation but not without reason. There has never been a case without repetitive head impacts (RHI) in the case histories. There is no model for the latency in CTE symptoms (it can take years to tens of years) relative to the case histories of RHI. What is particular and specific to CTE about these NFTs that McKee is seeing (and Dr. Bennet Omalu saw initially in Mike Webster’s brain) is their location around blood vessels (perivascular) and where they are found in the brain, initially but in all stages, in the sulci or grooves of the frontal lobe.
This is a good segue way back to McKee’s study where she shows the regions by stage where the NFTs are found.
McKee summarizes the number of NFTs by region of the brain on a zero to three scale by stage (I thru IV) as counted in a set field of view.
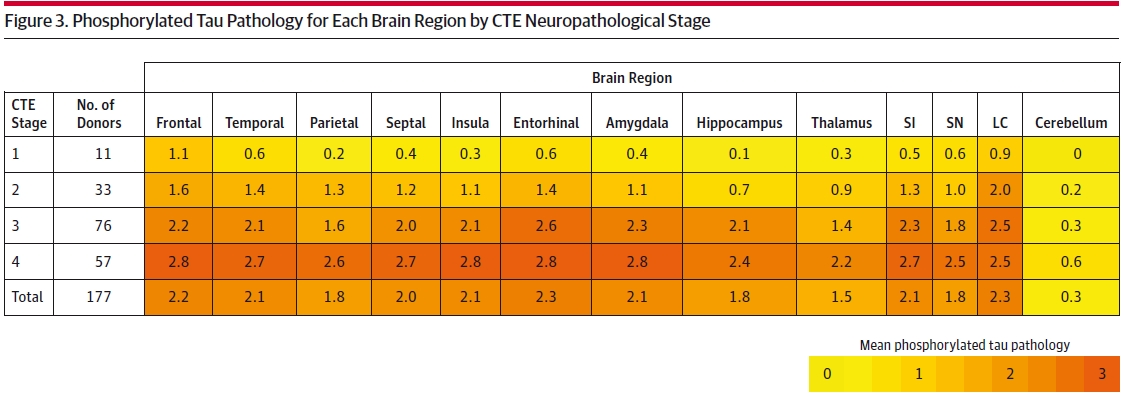
SI: substantia innominata, SN: substantia nigra; LC: locus coeruleus.
I don’t expect everyone is up to date on their brain regions. The colorization and the discussion, however, are clearly highlighting the observed progressive nature of the stages. This progression matches the reported progressivity in behavioral and cognitive symptoms in 96% of the retroactive clinical evaluations taken on the brains McKee studied. This calls for a bit of mapping of these symptoms to the regions of observed CTE. McKee does this with the caveat that some stage I and II subjects have symptoms that are not reflected in the regional NFT survey. She goes on to explain possible models for this. Mapping region to function is somewhat controversial but general precepts of function do match the clinical evaluation rollup of symptoms. The main take from this table however is that CTE is progressive.
McKee discusses the progressive model wrt the convenience sampling. Each brain is a snapshot of that person’s condition at time of death. Each clinical evaluation is not longitudinal over the course of the subject’s life. This is a major limitation of this study and the current means of looking at this disease. There are studies doing that. They are years out. Most importantly this puts an onus on McKee when she posits the progressive model.
This is a good time to reiterate that I am encouraging you to read these studies yourself. Questions have been raised but the data is what it is. The terms and discussions are very well written and referenced. At least you can see a hint of it here.
The discussion of Figure 3 does not mention the outliers here in rate of progression (the Parietal from I to II and the Hippocampus from I thru III.) All other regions except for the cerebellum show a declining rate of progression from Stage I thru IV. Stages I and II are markedly different and somewhat discontinuous from Stages III and IV. This is likely the reason for the newer categorization of Mild and Severe. If you take the approximate % difference from stage to stage you can see what I mean about the Parietal and Hippocampus…
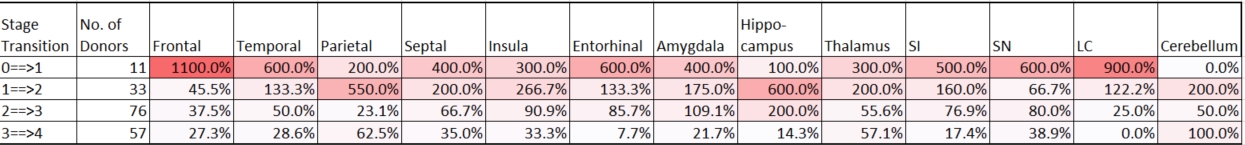
If CTE is progressive then the uneven progression is a mystery and clue to how it actually does this. McKee doesn’t talk about that and I don't think you can read too much into it. Looking at the actual data though makes you think about what is really going on with this disease. McKee's paper, on the other hand, is all about the what and where with a few hints of how and why.
Another take from this CTE by brain region figure and the concept of progression is that initially this disease seems to roost in the frontal lobe. Specifically, from the images and discussion, in the grooves of the lobe called sulci or sulcus for a single groove or fold. This is not where these exceeding long neurons I discussed previously happen to reside. Neurons come in all shapes and sizes. Brain neurons are different in structure and function from the cells most people commonly think about when, if ever, they do imagine the workings of the human brain.
If CTE is concussion/mTBI based it's odd that it wouldn't onset in the longer interleaving white matter. It doesn't. That said there are biomechanical models that show these sulci as more susceptible to trauma. MTBI and CTE don't have to be causative. Careful researchers are careful in not making that assumption. Careless talking heads, Frank Bruni and Malcom Gladwell, are careless doing exactly that. RHI is a sufficient precursor to explain CTE. All mTBI cases to date with CTE are a subset of RHI cases.
Last digression… Conflating CTE and mTBI is a story that the press continues to get wrong. MTBI may indeed be a precursor to CTE. Controlling mTBI, however, will not necessarily eliminate CTE risk. Concussions in and of themselves have very real short and long term risk. CTE is not necessarily one of them. Between 1/4 to 1/3 of us will have a concussion (mTBI) in our lifetime. The annual rate of concussion in high school has seen an increase of ~16% in the recent years (mileage may vary but this is from a 2011 11 year study in the American Journal of Sports Medicine). When CTE and mTBI are treated so light handedly in the media and the movies and on our board… we just start to lose focus on the science. Football suffers.
Rant complete…
Here is the break down by age and stage. There’s quite a bit more data in this table regarding neuropathological features and other diagnoses, like AD, Lewy body disease (LBD) and other neurodegenerative disorders. I’m skipping that for clarity and showing only the % Pure CTE data from that table.
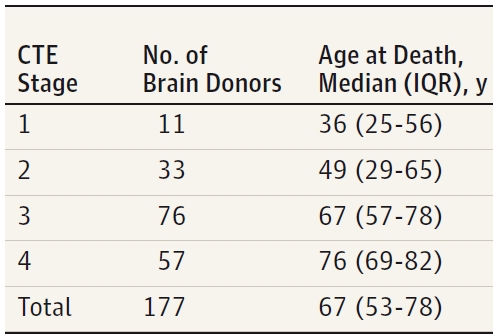

The big take here is that there is considerable crossover or comorbidity with AD, LBD and other diseases of the brain. This confuses the symptomatic signal and is another source of bombast and misinformation in fan response. The high comorbidity is perhaps a sign that the model for CTE is complex and integrative with other p-tau marked neurodegenerative disorders. The stark % of pure CTE found at all stages is still very high. The % in mild CTE cases is even higher. Bottom line… CTE is a demonstrable disorder. Before one dies, however, symptoms might be a sign of something other than CTE or worse… multiple pathologies. At-risk patients need to be met where they are at with a broad treatment plan addressing the treatable symptoms regardless of an attribution of cause to previous TBI.
One more table and I'm done. I will skip the Stern study.
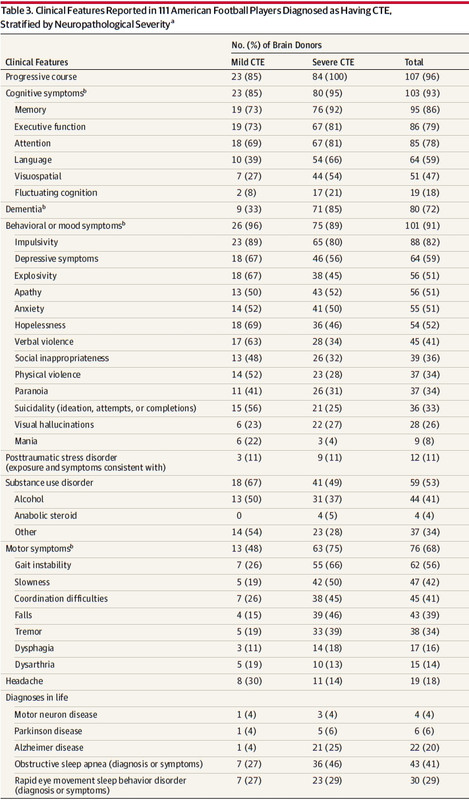
Here you get a feel for the difference between mild and severe presentations which is confounded by the age distribution from the second Table. The cognitive decline with age and severity are explained in the pathology. But why, if CTE does progress, does the presentation morph away from the behavioral symptoms to a cognitive presentation going from the mild stages I and II to the severe III and IV? Why does the substance abuse decline?
Just as McKee explained with the behavioral symptoms not having an analogous pathological correlate in the Mild stages their relative remission in severe staging is not correlated to profound NFT density and progress either. Behavior doesn't track with NFT density or severity. That would seem to me to be another take on the Hernandez case wrt to this study. But as I mentioned before… if his slides are truly exceptional… no study is going to shed light on his premeditation of murder or his choice to carry it out.
The other study “Age of first exposure to American football and long-term neuropsychiatric and cognitive outcomes” in the Journal Translational Psychiatry was just published last week and had considerable press. This is a completely different work from the previous study by Stern on the Age of First Exposure (AFE) and CTE, which I blogged about two years ago. Since that study was done the NFL sponsored their own study that did not replicate those results.
It’s funny to compare the NFL study to Stern's work. That may be my own special interpretation of funny. That is the post this diary should have been and was when I started this last week. When I did start I quickly realized there was other stuff that hasn't been blogged that needed coverage first. There is a ton of post-worthy material coming out all the time. There is little time to blog about it. I’m over 3000 words so I will skip that comparison if only to make another.
Tom Brady and Aaron Hernandez, let's compare and contrast. Stern’s and McKee’s studies show AFE and CTE and behavioral and cognitive decline. Though you wouldn’t know it from Frank Bruni’s editorial Brady didn’t play youth Football. Aaron Hernandez, on the other hand, received a life time award by Pop Warner in 2013 just prior to his arrest. The two cases couldn’t be any more different with respect to AFE as an indicator.
The NYT didn’t editorialize the Hernandez story. They did however choose to put it on the front page. Fan interest drives that as much as editorial choice but the implication is clear. CTE can make you a murderer. That is nowhere near what the science is showing. Aaron Hernandez is an n of 1. The front page treatment is sensational, pandering and unfair.
There is absolutely zero evidence Tom Brady has symptoms or precursors of CTE. As McKee mentioned … no generalizations can be made from her study. It doesn’t do anyone any good to assume one athlete or another has the disease until they shows signs or are conclusively proven to have had it, for the time being at least, after they pass away. As I have blogged before, the real attack on football is the conflation of CTE with concussions. The hit piece on Brady is that in spades. It could do more harm if Brady is later shown not to have CTE…which at this point, in my mind at least, seems likely. It is a head game that no one wins.
It's five days since I started this diary. My cat has already deleted it twice crawling across my laptop. She likes the heat. She obviously wants a part of this post but I will skip the LSA cat picture.
Happy Improvement Week. Go Blue!
Edit - for a broken link and some typos.
September 29th, 2017 at 6:13 PM ^
September 29th, 2017 at 7:45 PM ^
A) Excellent write up
B) As an epidemiologist with a strong background in toxicology, to me the most interesting issue is that of exposure and dose. Getting at a better metric for the exposure than simply years played and position is a fascinating challenge. Likewise, understanding the true prevalence of CTE in the general population is quite an undertaking.
C) With regards to the CTE + Alz issue, "mixed dementia" or Alz occuring simultaneously with Lewy bodies and vascular dementia, is similar. I'm not entirely sure what the cycle of chicken and egg is with those diseases, but I'd imagine the complexity is similar to that of CTE + Alz/Parkinsons.
September 29th, 2017 at 11:44 PM ^
the convenience sample pretty much kills any statistical power? Then again the goal is not to calculate prevalence or incidence, more to understand what is going on with the sample population?
Kind of like where researchers review the medical records of only hospital patients with a certain condition or only certain populations where the condition is understood to exist?
September 30th, 2017 at 11:03 AM ^
have to edit that. By definition, a convenience sample has no statistical power, and is in no way a scientific sample, and cannot be generalized to the target population.
October 1st, 2017 at 12:54 AM ^
YOLT...your take is pretty much dead on to the limit of my understanding. This is a study describing the disease but not the prevalence. I had a draft of this diary that got into that (damn cat!)
The hit rate, however, wrt NFL players in this convenience sample is so high as to lay the ground work for a follow up study on prevalence and incidence that, if done carefully, would have statistical power. Even then it would need follow up pathology to confirm.
The league and the players union are not doing that. I am not sure if that would help at this point. It seems the public vis a vis the media have already determined that number to be very high.
We don't have a model for the latency or progression of CTE. The conflation of mTBI with CTE gives a false sense of containment to the public. Finding the actual number wouldn't serve the NFL or the players union at this point in my opinion. The players union settlement sold the younger players and amatuers down the river.
Former players have their own brains to sample. This study is very helpful to them to differentially sort that out. If they are younger, self medicating and suffering from mood or behavior symptoms they might suspect mild CTE. If they are older and suffering cognitive symptoms they should be wary of severe CTE.
There is no treatment other than matching symptoms to presentations. It's early on in that regard.
Scans are another area of research I wrote in and then deleted. Functional MRI and other techonolgies are pretty interesting... but unfortunately can add up to kookery if not done well. This guy is an example of that. If you click thru you see how everyone is so willing to believe a scan can see stuff it just can't. That said there is a ton of interesting work being done.
Yada yada... I agree.
October 3rd, 2017 at 2:18 PM ^
2 points here:
1) Convenience sampling indeed has severe limitations, as the authors clearly state in the JAMA article. Statistics from studies are typically inferential - that is, they infer something about a risk or an association to a much greater population of interest.
HOWEVER: This convenience sample is very different in that we are discussing a much smaller population of interest: NFL players. The nyt review does a nice job of not missing the forest for the trees here. They note that even if every other NFL player who had died since the study began had tested negative for CTE (which of course wouldn't be the case), then the overall CTE rate would still be extremely high at about 9%. This means that we can infer that the CTE rate for NFL players that have died during this period is between 9% and 99%, both of course are extremes and therefore extremely unlikely. Even so, the 9% lowest possible rate would translate to an average of about 5 players on each NFL roster.
2) I applaud the OP's attempt to present the scientific merit of the new research. We still have a long way to go in science writing, and when the general population does not even have access to articles like this (in fact, the reproduction of tables in this entry are not strictly allowed for public dissemination), it will be a long road that I don't see us completing any time soon.
October 4th, 2017 at 1:03 PM ^
perspective.
The careful authors of the studies I reviewed here don't speculate on actual prevalence of CTE in the NFL. The careless NYT does. If you go by their numbers.... well that is how you ride.
I am a subscriber and read the NYT opinion, business and news section every day and have since my Michigan days. So far that has got us into a few needless wars and other debacles that good journalism could have avoided. I expect more from our country's ersatz paper of record. (I love the NYT by the way. I cut my teeth on it every day.)
I also ran some figures when writing this diary up. I decided not to include that since I honestly don't want to lose the forest for the trees.
I take exception to the NYT 9% to 99% numbers. They mention the ~1300 NFL players who died since they started microtoming brains at Boston Univ and take their 9% (110) from that number. That doesn't count those still living. Including them for the mild sample years that is ~9500 living and dead (going back to my favorite CFB year 1997.) For the severe sample life span that would be ~22000 players going back to 1967. That would give a low ball weighted average hit rate of 0.37%. That is 1/27th what the NYT listed and ~1/300th their high ball number. So yeah... numbrz.
As you metioned, this sampling doesn't allow population estimates. We need to be careful about that. Science doesn't give answers without a healthy dose of skepticism. People are looking for answers and the media is giving them without substance.
You are so right about science writing and I might take it further science. The best and most authoritative ideas of science are so unfortunately termed theories. Discoveries are housed in elite journals that my public library can't even afford to buy.
I think this is fair use but I take your critique. Most people "read" these papers by spending the majority of their time looking at the charts, tables and figures and skimming the methods, results and discussion. I did not quote the written sections at all. That, along with the references, is the heart of the paper.
I regretfully don't have a reference for the excellent timeline which I picked out of an editorial. I had it annotated and credited, but as I mentioned someone deleted all but the first couple paragraphs. Embarassingly I couldn't ctl-z it back. I apologize to that author and will add credit back once I find it again which I will.
McKee's study is available to anyone with the proper (improper?) URL. They just need to snoop it. My initial link is to the pay site. If people don't want to snoop, most libraries are consorted to allow access.
One of my favorite people (I am going to get slammed for this) said "You win with people." The forest here are the players living and dead who are being studied without true access to the results. That was the intent here. If fair use is questioned... that is the use I would term fair.
This sounds defensive. It's not. I disagree. Go Blue!
September 30th, 2017 at 9:19 AM ^
September 30th, 2017 at 3:45 PM ^
September 30th, 2017 at 7:11 PM ^
October 1st, 2017 at 2:34 PM ^
the state of the science is way different than the media reports would have you believe--same for the vast majority of commenters on this board. It seems that people have already concluded that Football = CTE and that is simply not proven (yet) in the science. I wish some of the same people that respect the science on climate change and other issues would actually apply the same standards to CTE, but they are not.
October 1st, 2017 at 9:40 PM ^
Impressive scholarship.
I hope many people read this diary.

Comments